Introduction
Granular cell tumors (GCTs), previously known as Abrikossoff tumors [1,2], are benign neoplasms of Schwann cell origin. These rare tumors account for less than 1% of all soft tissue tumors. Although the most commonly involved region was reported to be the oral cavity, especially the tongue [2], this is not the only region affected [3]. GCT of the auricle is exceptionally rare, with only one case reported to date [4].
Cutaneous GCT (cGCT) mostly presents as a painless, slow-growing, solitary nodule with intact skin coverage. Therefore, this tumor type is not always considered in the differential diagnosis of a lesion. Due to their subtle clinical presentation, these tumors are often misdiagnosed [5]. Here, we report a rare case of auricle GCT, which illustrates that histopathological examination is essential for correct diagnosis.
Case Report
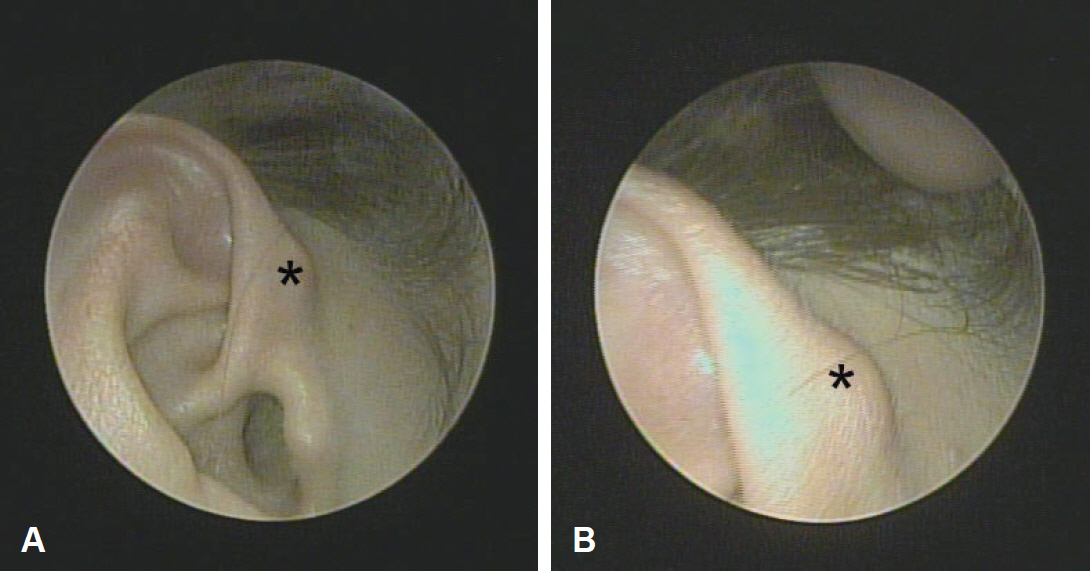
A 47-year-old woman presented with a painless mass in the right auricular helix that had been gradually enlarging over a year. Physical examination revealed a 1-cm-sized, soft, subcutaneous nodule with normal overlying skin (Fig. 1). She had no history of auricular surgery or trauma.
The initial clinical differential diagnosis included epidermoid cyst, adnexal tumor, such as pilomatricoma, dermatofibroma, neurofibroma, lipoma, and schwannoma. The surgical excision, which included the mass and the overlying skin, was performed under local anesthesia. Grossly, the mass was 0.8 cm in diameter, pinkish, and firm, with a well-demarcated capsule (Fig. 2).
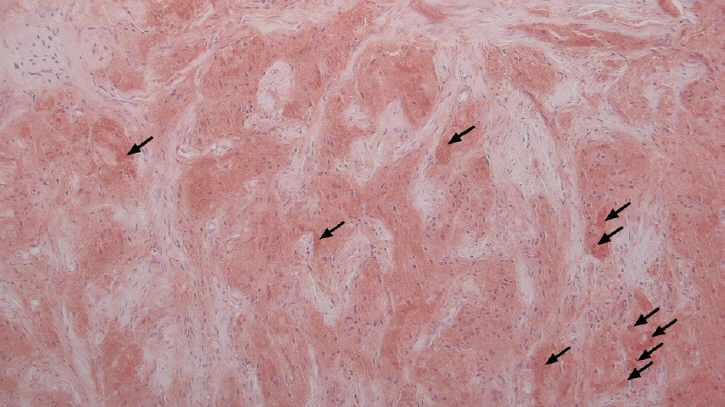
A histopathologic examination showed that the dermis was infiltrated with cells containing granular cytoplasm without cytologic atypia. In the subcutaneous fat layer, the borders were irregular and infiltrating. There were irregular entrapments of adipocytes by the granular cells. These eosinophilic granules were periodic acidŌĆōSchiff (PAS)-positive. Eosinophilic ovoid bodies were surrounded by a clear halo (Fig. 2).
Immunohistochemical staining revealed that the tumor cells were positive for S-100 (Fig. 3) and neuron-specific enolase (NSE), but negative for epithelial (cytokeratins), melanocytic (HMB-45), muscle markers (╬▒-SMA) (the data was not included). This staining pattern was also suggestive of a Schwann cell origin.
Based on the immunohistochemistry results and morphological features of our case, we excluded possible diagnoses of epidermoid cyst, adnexal tumor, dermatofibroma, and melanoma. Based on the distinctive histopathological and immunohistochemical features, the patient was diagnosed as having benign cGCT. No recurrence was observed during a 24-month follow-up period post-excision.
Discussion
The origin of GCT has long been debated. GCT was initially thought to be of myogenic origin, and was named granular cell myoblastoma [1,2]. However, based on electron microscopy studies that revealed the presence of axonal structures, and immunohistochemical studies that demonstrated S-100 positivity, GCT is now thought to have a Schwann cell origin [6].
GCTs do not have a significant predilection for any age, race, or sex. They mostly present as a painless, slow-growing, solitary, firm, dome-shaped mass in the subcutaneous tissue, with overlying normal skin. Cases of congenital GCT and familial cases have been reported to be associated with multiple lesions [5].
GCT pathogenesis is unknown, but there have been reports of GCTs that have developed at sites of previous trauma or manipulation. However, occasionally, reactive changes after injury can also mimic a GCT [7].
cGCTs can be clinically classified into two subtypes (verrucous and smooth cGCT). Externally, cGCT often has a verrucous appearance that is overt hyperkeratosis in the overlying skin. Histologically, these findings correspond with pseudoepitheliomatous hyperplasia. On the other hand, apparently normal epidermis is found in smooth cGCT as in our case study.
As cGCT does not present with any specific clinical features, this tumor is seldom considered in the differential diagnosis of lesions with this appearance. GCT is mostly diagnosed via histopathological examination, with immunohistochemical staining [5]. Histopathologically, the tumor cells possess abundant, pale, eosinophilic, and granular cytoplasm.
Abundant cytoplasmic lysosomes are responsible for the tumor cellsŌĆÖ granular appearance. The cytoplasmic granules were found positive for PAS staining, and for S-100 protein, NSE, CD-57, and vimentin following immunohistochemistry [8]. The reason for the granular changes is unclear, and a reactive or degenerative response has been suggested. The electron microscopy observations revealed that the granules were degenerated myelinated axons [7,9].
The desmin staining pattern in GCTs can be useful for distinguishing these neoplasms from smooth muscle tumors. In GCTs, cells are usually negative or a few cells are weakly positive for desmin, whereas leiomyomas are strongly and diffusely positive for this protein.
The clinical differential diagnosis of cGCTs includes lipoma and adnexal tumors, such as dermatofibroma, neurofibroma, and schwannoma. Furthermore, basal cell carcinomas, melanomas, melanocytic nevi, leiomyomas, leiomyosarcomas, dermatofibrosarcomas, rhabdomyomas, angiosarcomas, and ameloblastomas have granular cell variants. These can be differentiated from GCTs via immunohistochemical studies [5]. S-100-positivity in GCTs helps to exclude alveolar soft part sarcomas and rhabdomyomas.
In our case, the tumor was composed of typical cells with a granular eosinophilic cytoplasm, which stained positive for S-100, NSE. Due to these distinctive immunohistochemical features, our patient was diagnosed of GCT. Furthermore, we could exclude possible diagnoses of dermatofibroma, melanoma, and malignant neural tumor.
GCTs are mostly benign; however, malignant transformation occurs in 0.5-2% of cases [10]. Metastasis to the regional lymph nodes, lungs, and bones are most commonly reported. It was challenging to predict malignant behavior at the time of diagnosis [5]. Malignant GCT can be differentiated from benign GCT by its larger size (>4 cm) and rapid growth, and histopathologically, by a vacuolar nucleus with an enlarged nuclear body, increased mitotic activity, a high nuclear-to-cytoplasmic ratio, necrosis, spindle cells, and pleomorphism [11]. Without these features, the tumor can be considered to be benign.
Furthermore, even benign GCTs can show locally invasive features, such as local recurrence and metastases. Benign GCTs have a recurrence rate of between 15% and 50%, if the tumor is not completely excised [7,9,12,13]. As in this case, GCT could have irregular, infiltrating borders. Moreover, it has been noted that histologically, benign-appearing tumors can metastasize [14,15]. Thus, GCTs do require wide excision, leaving clear margins. Radiotherapy and chemotherapy have not been recommended in the recurrent or malignant cases [5].
In conclusion, this case may remind otologists that GCT should be considered in the differential diagnosis of a solitary nodule at the auricle. Because benign-appearing GCT can metastasize or recur, complete excision is recommended. Subsequently, histopathology should rule out the signs of malignant change, even though this is rare.