Introduction
Sensorineural hearing impairment (SNHI) stands as the leading sensory deficit in children, affecting approximately 1.9 per 1,000 live births [1], with an additional 1 per 100 school-aged children presenting with late-onset SNHI [2]. Genetic factors contribute to more than 50% of SNHI cases, with more than 200 genes identified to date [3]. For children with mild or moderate SNHI, hearing aid fitting is the primary intervention. However, in cases of profound SNHI, cochlear implantation becomes mandatory.
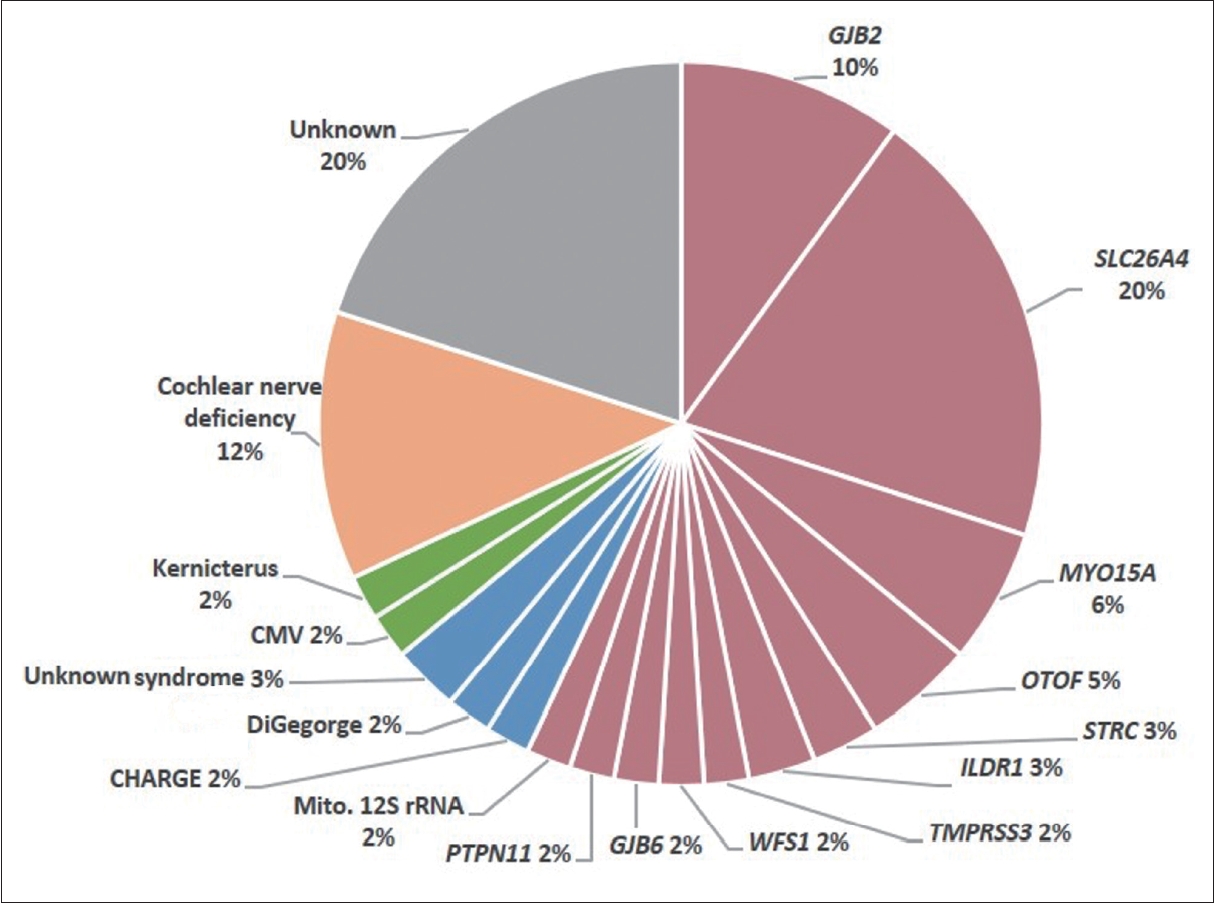
Advances in genomic medicine, particularly next-generation sequencing (NGS), have significantly facilitated the integration of genetic examination into clinical practice. NGS-based genomic sequencing has gradually replaced traditional genetic testing and has become the main diagnostic tool for pediatric SNHI [4-7]. In combination with imaging and virological studies, NGS allows etiologic identification in approximately 80% of pediatric cochlear implant (CI) candidates [8-10]. This includes approximately 50% of cases with non-syndromic SNHI, approximately 10% with syndromic SNHI (e.g., Waardenburg syndrome), approximately 10% with acquired SNHI (e.g., congenital cytomegalovirus infection), and approximately 10% with inner ear malformations (e.g., cochlear nerve deficiency) (Fig. 1) [8,9].
Management of pediatric SNHI involves sequential clinical decisions that take into account the severity and progression of hearing loss. The potential for progressive hearing loss prompts consideration of the need for cochlear implantation and the optimal timing of such intervention. If cochlear implantation is subsequently deemed necessary, an assessment of the potential benefits to the patient becomes paramount. The final decision to proceed with surgery involves careful selection of the most appropriate device or electrode for the individual patient. Genetic information can provide critical insight into these decision-making processes.
Tracking the Evolution of SNHI to Determine Optimal Surgical Timing
Genetic diagnosis plays a pivotal role in unraveling the natural progression of SNHI and determining the ideal timing for cochlear implantation, closely aligning with the clinical course. Different pathogenic variants are associated with different patterns of SNHI progression; for example, GJB2 mutations are associated with stable or slow progression [11-13], whereas SLC26A4 mutations are associated with fluctuating SNHI [14-16].
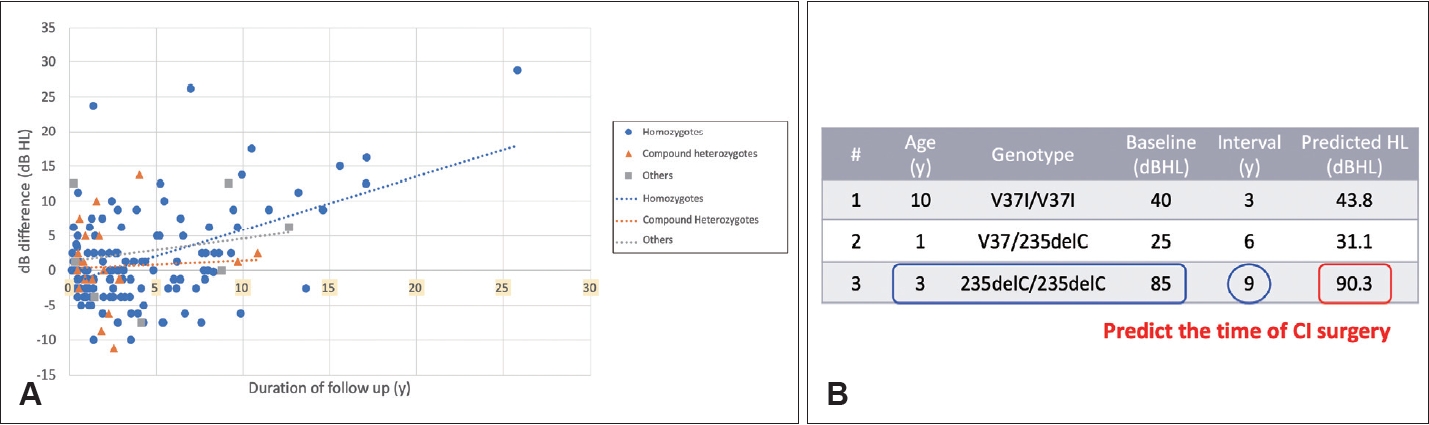
The close relationship between genetic diagnosis and clinical progression allows prediction of the timing of CI surgery. In a previous study, we formulated a prediction model for GJB2-related SNHI [17]. Our results showed the severity of hearing loss in individuals with pathogenic GJB2 variants correlated with their genotypes and baseline hearing levels. On average, the rate of hearing loss deterioration was approximately 0.55 dB per year, as shown in Fig. 2 [17]. This observation was corroborated in a recent study that reported a rate of deterioration of approximately 0.3 dB per year in patients with pathogenic GJB2 variants [18].
This predictive model serves as a tool to estimate when a patient’s hearing may deteriorate to the point where cochlear implantation may be required. For example, consider a 3-year-old child homozygous for the GJB2 c.235delC variant (Fig. 2B, case #3) with an initial hearing level of 85 dB HL. Given that the eligibility criterion for cochlear implantation is 90 dB HL, the model predicts that approximately 9 years later, the child’s hearing loss is likely to reach the 90 dB HL threshold, making him or her eligible for insurance coverage of a CI.
Predicting the Potential Benefits of CI in Patients With Identified Genetic Causes
Genetic insights into CI success
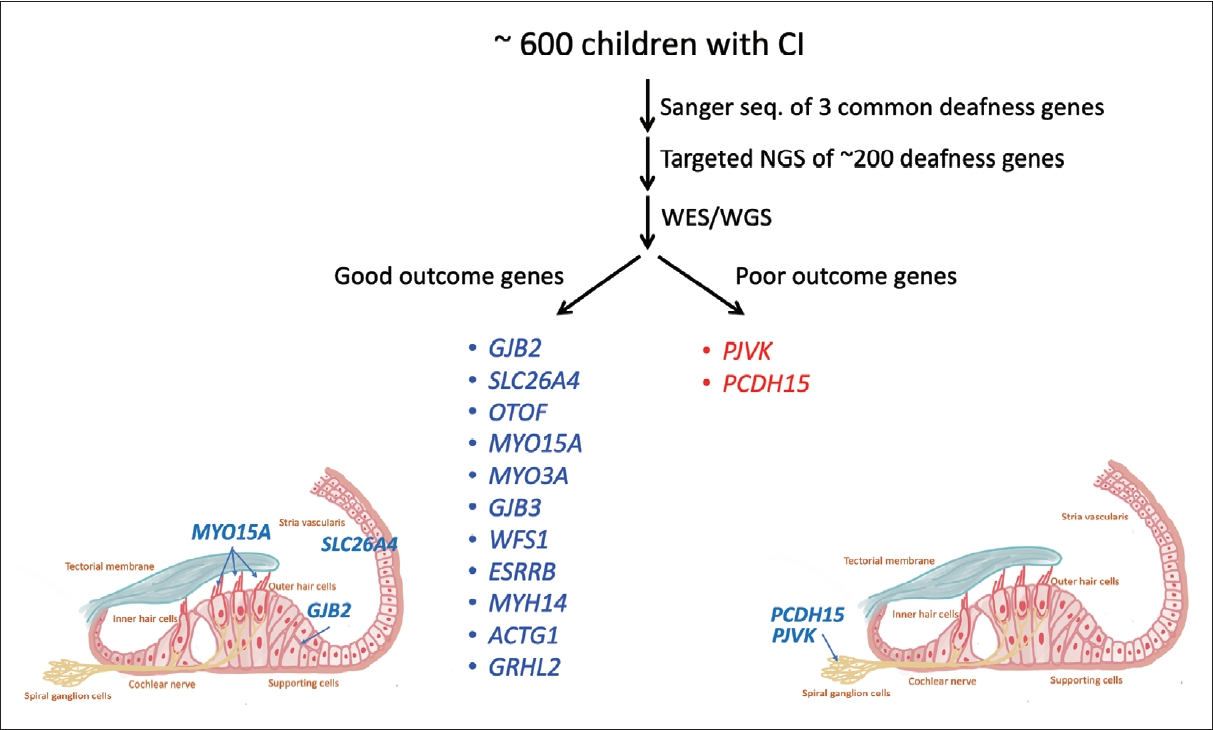
In addition to predicting disease progression, genetic diagnosis plays a critical role in guiding treatment decisions, particularly those related to cochlear implantation. Our extensive genetic studies involving more than 600 CI patients in Taiwan have revealed clear associations between genetic mutations and CI outcomes (Fig. 3) [8,19-21]. Pathogenic variants in most genes, such as GJB2, SLC26A4, and MYO15A, are associated with favorable CI outcomes, likely due to confined pathology within the inner ear [8,19-21]. These findings are consistent with other series [10,22] and have important clinical implications. Identification of pathogenic variants in these genes serves as a reliable predictor of CI outcomes, expediting the decision-making process for otologists, audiologists, geneticists, and patients.
Conversely, pathogenic variants in genes such as PJVK and PCDH15 are associated with less favorable CI outcomes [23], likely due to involvement of spiral ganglion neurons [24]. Notably, pediatric patients with pathogenic PJVK and PCDH15 variants presented with clinical features indistinguishable from those of other typical pediatric CI recipients, underscoring the need for genetic testing for accurate diagnosis [23].
Auditory neuropathy spectrum disorder
Genetic diagnosis is particularly valuable in the management of auditory neuropathy spectrum disorder (ANSD), which constitutes approximately 10% of pediatric SNHI cases [25]. The heterogeneous pathologies of ANSD range from inner hair cells to the auditory cortex [26]. Our previous study highlighted that acquired factors such as prematurity and kernicterus are the predominant contributors to pediatric ANSD, while genetic factors account for approximately 1/5 of cases, with pathogenic OTOF variants being the primary genetic cause [27].
Depending on the location of the lesion, ANSD is classified as pre-synaptic or post-synaptic. In general, pre-synaptic ANSD is associated with positive CI outcomes, whereas post-synaptic ANSD is associated with less favorable CI outcomes [26]. The exact location of the pathology is critical. For instance, OTOF variants primarily affect the synapse, leaving the neurons intact for CI function. Consequently, patients with pathogenic OTOF variants exhibit robust electrically evoked compound action potential responses during surgery and excellent hearing and speech performance after surgery [28]. Therefore, early cochlear implantation is recommended for ANSD patients with pathogenic OTOF variants.
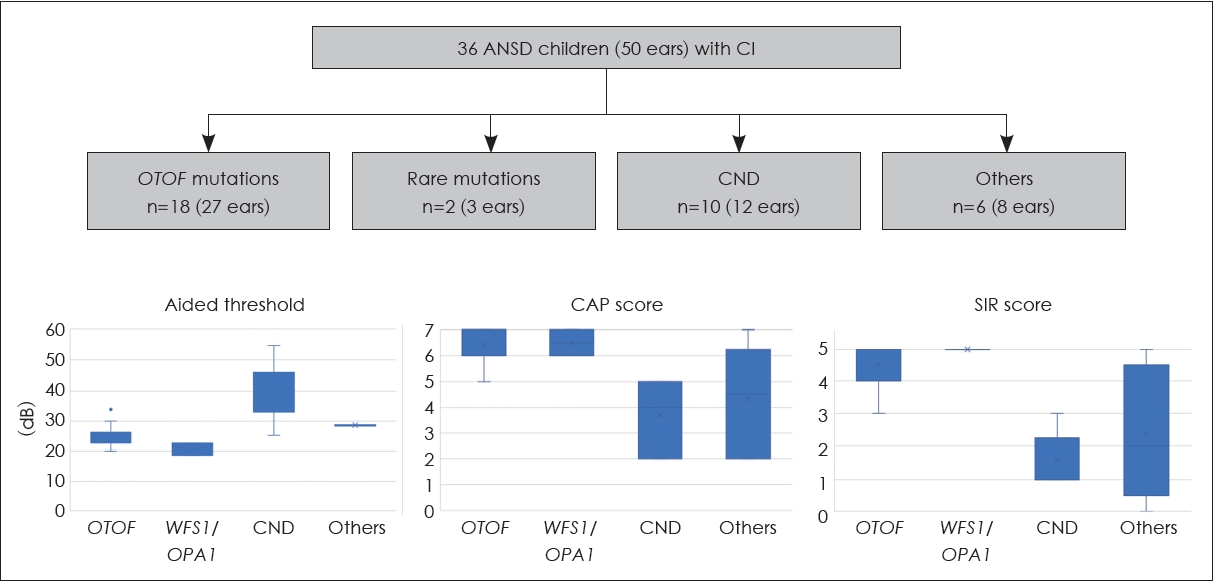
In a recent study at the National Taiwan University Hospital, we analyzed CI outcomes in 36 ANSD patients, including 18 with pathogenic OTOF variants, 2 with rare pathogenic variants in the OPA1 and WFS1 genes, 10 with cochlear nerve deficiency (CND) and 6 without a definite diagnosis (Fig. 4) [29]. Aided behavioral thresholds showed improvement in all patients after surgery, although they were higher in the CND group. In particular, patients with OTOF, WFS1, and OPA1 variants showed favorable CI outcomes, as evidenced by good categorical auditory performance and speech intelligibility rating scores. In contrast, patients with CND had suboptimal outcomes, highlighting the importance of identifying ANSD etiologies prior to surgery for prognostication and counseling.
Unilateral or asymmetric SNHI
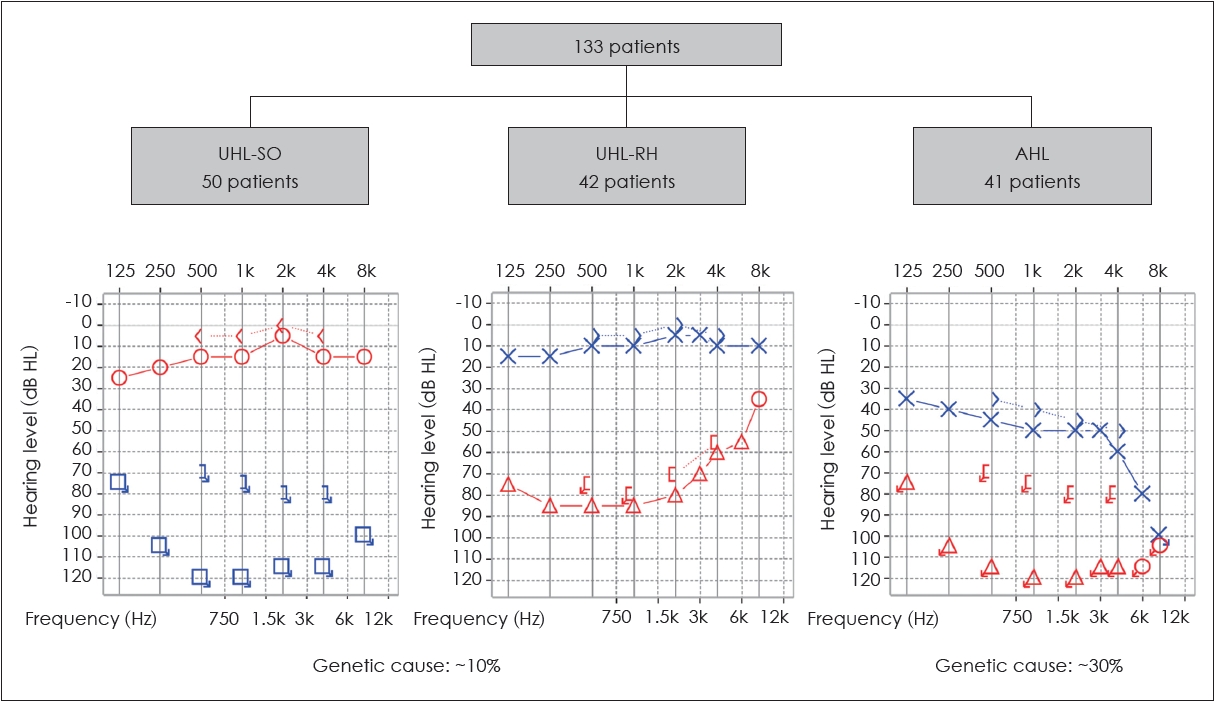
In addition to ANSD, genetic information can guide clinical decision-making for patients with unilateral or asymmetric SNHI, a condition for which cochlear implantation has emerged as a viable treatment option [30,31]. However, similar to bilateral SNHI, the etiologies of unilateral or asymmetric SNHI are remarkably diverse. In our recent studies, we analyzed the genetic basis in patients with unilateral or asymmetric SNHI. Our results showed that genetic causes could be identified in about 30% of patients with asymmetric SNHI (Fig. 5) [32]. Notably, genetic causes were also identified in approximately 10% of patients with unilateral SNHI [33], which was traditionally thought to be less likely to have a genetic origin. Specifically, pathogenic variants associated with Waardenburg syndrome were found to be a significant contributor to unilateral severe-to-profound SNHI [33]. This finding has significant clinical implications by suggesting a favorable candidacy for cochlear implantation, as the pathology of Waardenburg syndrome primarily affects the stria vascularis while leaving the cochlear nerve intact [34].
Selecting CI Devices/Electrodes Tailored to Patients With Specific Genetic Mutations
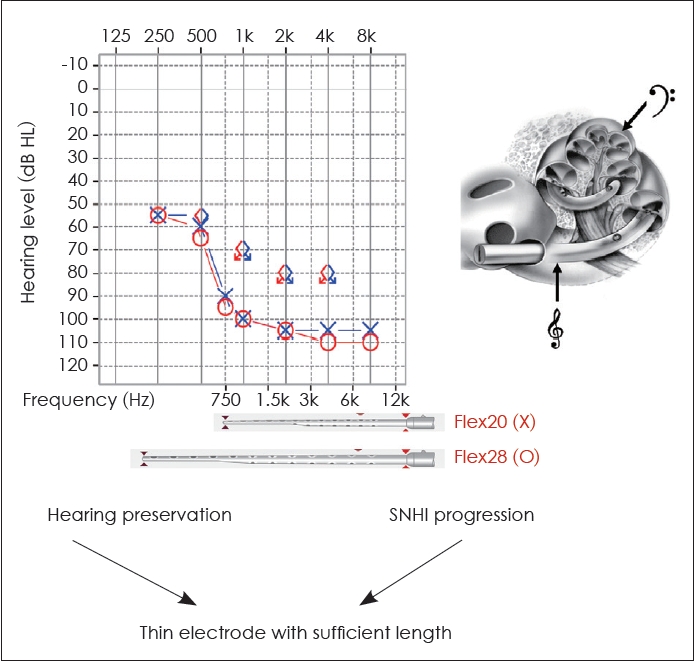
Genetic diagnosis plays a critical role in enabling clinicians to make informed decisions about electrode selection for cochlear implantation. Preservation of low and mid-frequency hearing has been observed in the early stages of certain genetic hearing impairments, such as pathogenic variants in the genes MYO15A [35,36], CDH23 [37], or TMPRSS3 [38]. However, a significant proportion of this residual hearing tends to gradually deteriorate over time [35-38]. Therefore, for individuals with MYO15A, CDH23, or TMPRSS3 variants seeking cochlear implantation, electrode selection must consider both the preservation of residual hearing and the progression of SNHI, with a thin electrode of sufficient length being the preferred choice. For example, when using MED-EL electrodes, the Flex20 electrode should be avoided. While the Flex20 may be beneficial for preserving hearing, it cannot cover the low-frequency range if residual low-frequency hearing is lost in the future. In such cases, the Flex28 electrode would be a more appropriate choice for the patient (Fig. 6). Thus, incorporating genetic findings into the selection of CI devices/electrodes allows for a more personalized, patient-specific auditory treatment.
Conclusion
In summary, our research over the past two decades underscores the critical role of genetic information in guiding clinical decisions related to cochlear implantation. The impact of genetic knowledge extends to multiple aspects of patient management, allowing the tracking of SNHI evolution to determine optimal surgical timing. In addition, the predictive power of genetic information is proving valuable in assessing the potential benefit of CI for diverse patient populations, including those with ANSD or unilateral SNHI. Finally, our findings highlight the importance of tailoring CI electrode selection based on specific genetic mutations, providing a more personalized approach to improving the efficacy of cochlear implant interventions.